
Ab initio Model for Structure-Bioactivity Relationship
Advanced Materials Characterization
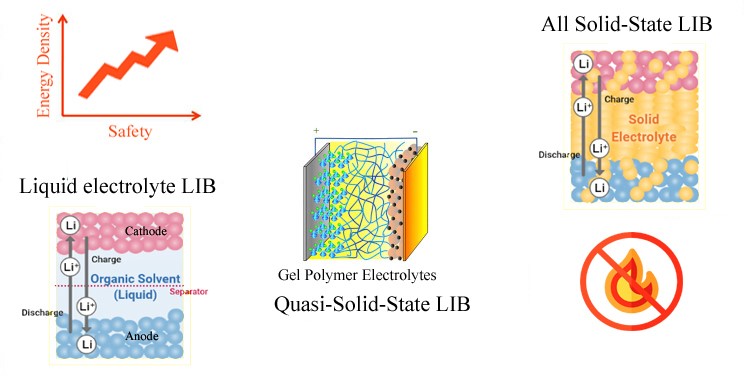
Lithium-ion batteries (LIBs) are thriving because of high energy density, high efficiency, and long life. These unique aspects make LIBs as the power sources of choice for sustainable transport since they can effectively guarantee the progressive spread of full hybrid electric vehicles (FHEVs), plug-in electric vehicles (PEVs), and battery electric vehicles (BEVs) at high levels. They are also expected to find an outstanding role in power-storage systems in renewable energy plants. A conventional LIB is composed of cathode, anode, membrane separator, and liquid electrolyte (LE). In contrary, an all solid-state battery (ASSB) uses a solid-state electrolyte (SSE). The substitution of SSE for organic LE lifts the risk of flammability at elevated operation temperature when the battery has been accidently degraded or damaged (through impact, overcharge, crush, short-circuit, etc.). Hence, works on high performance SSEs for LIBs have caught tremendous attention. According to WOS search results, the number of publications on SSE-LIBs has dramatically increased from about 500 in 2010 to over 2500 in 2021.
Existing SSEs can be categorized into 3 groups: (i) inorganic SSE, (ii) composite SSEs, and (iii) solid polymer electrolytes (SPEs). The main goal of this research is to improve the performance of the SSE-LIBs to the LE-LIBs. There are several attempts reported to engineer SSE material to increase the ion mobility. Theoretical approaches provide a cost-efficient process to understand the microscopic ion transport mechanism, complement experimental analysis, screen and predict properties. The theoretical calculation has been used to predict Li+ ion conduction process, but most of the reports could not explain the actual mechanism and predict conduction mechanism properly.
Theoretical analysis has been practiced in every aspect of the battery material to understand and link up various aspects like diffusion in electrode or electrolyte, an electrical response, interfacing between electrode and electrolyte, the effect of charge discharge process on the battery structure etc. There are mainly two forms of theoretical approaches practiced on the battery materials: interatomic potential based classical analysis (mainly molecular dynamics (MD) method) and electronic structure methods (mainly density functional theory (DFT) method). The first approach is based on the potential model developed on the classical understanding of the interaction between different atoms and calculating the total energy. The MD route consists of solving Newton’s equations of motion to predict time dependent trajectories and can be employed in a large ensemble of particles in a finite temperature and pressure. Applying the method on Li ion material we can extract the information of ion mobility in certain operation environment and compare different characteristics like diffusivity and conductivity and closely observe the behaviors which is not yet possible in the most sophisticated equipment available up to now.
In our group, Li+ migration barrier in the crystal is analyzed using electronic structure DFT model and ab initio MD technique is used for the case of an amorphous structure.